诱导效应与共轭效应
你有没有想过,为什么同样是酸,盐酸(HCl)的酸性远比醋酸(CH₃COOH)强?而在醋酸的基础上,把离羧基最近的那个碳上的氢换成氯,得到氯乙酸(ClCH₂COOH),酸性又会立刻变强许多?仅仅换了一个原子,酸性就发生了显著变化,背后推动这个变化的正是今天要讲的主角——电子效应。
有机分子不是一堆“静止”的球棍模型,分子中的电子时刻处于运动与偏移之中。当一个原子或基团靠近分子骨架,它会通过化学键“悄悄”地把附近的电子拉过来,或者推出去,进而影响整个分子的电荷分布,改变分子的酸性、碱性乃至反应活性。这种微妙的“电子搬运”,在有机化学中被细分为两类:沿着单键传递的诱导效应,以及在共轭体系中大范围流动的共轭效应。
诱导效应:沿键传递的电子偏移
什么叫“电子被拉偏了”
在物理课上学过,当两个电荷量不同的物体靠近,较强的那个会把电场线“拉向”自己一侧。分子中也有类似的事情发生。
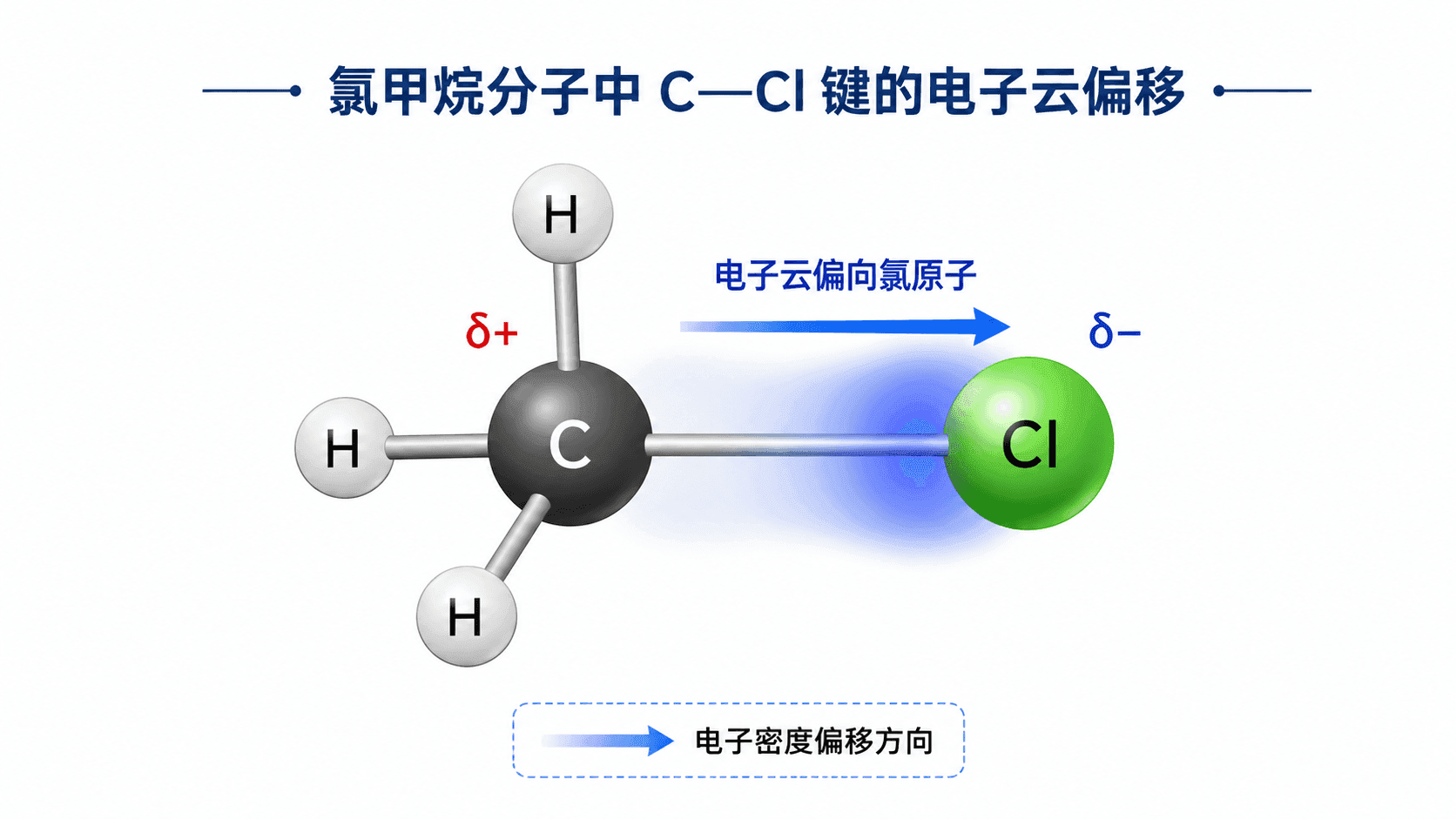
以氯甲烷()为例。氯(Cl)的电负性为 3.16,碳(C)为 2.55,氯的“吸电子能力”比碳强。连接 C—Cl 键中,共用电子对被氯原子偏向吸引,碳原子上的电子云密度相对降低,略显正电性(),氯原子那端电子云密度相对增加,略显负电性()。
这就是诱导效应(Inductive Effect,简称 I 效应)的基本含义:由于原子或基团的电负性差异,电子云沿着化学键方向发生整体偏移的现象。
吸电子基团与供电子基团
不同的原子和基团,吸引电子的能力各不相同。按照对比氢原子(H)作为基准,把比氢更“抢”电子的归为吸电子基团(-I 效应),把比氢更“推”电子的归为供电子基团(+I 效应)。
对于卤素(F、Cl、Br、I),电负性从 F 到 I 依次降低,吸电子诱导效应的强弱顺序为:
对于烷基,碳链越长、支链越多,供电子能力越强,叔丁基()的供电子能力比甲基()更强。
诱导效应沿键“衰减”
诱导效应最关键的一个特点是:它是沿着 σ 键传递的,而且随着距离增大,效应会迅速衰减。
一般来说,经过三到四根键之后,诱导效应几乎可以忽略不计。
用一根碳链来感受这一点:
氯对第 1 个碳的影响最强,对第 2 个碳已明显减弱,到第 3、4 个碳时影响几乎感知不到。
诱导效应的传递方向是固定的:从电负性弱的原子流向电负性强的原子。整个过程只是电子云的“整体偏移”,并非电子的真实转移,所有键仍然是共价键,只是极性发生了变化。
诱导效应对酸性的影响
回到最开始的问题:氯乙酸为什么比乙酸酸性强?
乙酸(醋酸)失去质子后,剩下的乙酸根负离子()需要稳定存在,才能让失去质子的过程更容易发生。氯乙酸中,氯原子通过诱导效应持续“抽走”邻近碳上的电子,这一效应传递到羧基部分,使羧基的 O—H 键的极性增大,氢更容易以质子()的形式离去;而且氯乙酸根负离子中,氯继续吸引负电荷,使负离子更分散、更稳定,从而促进电离。
三种乙酸衍生物的酸性对比:
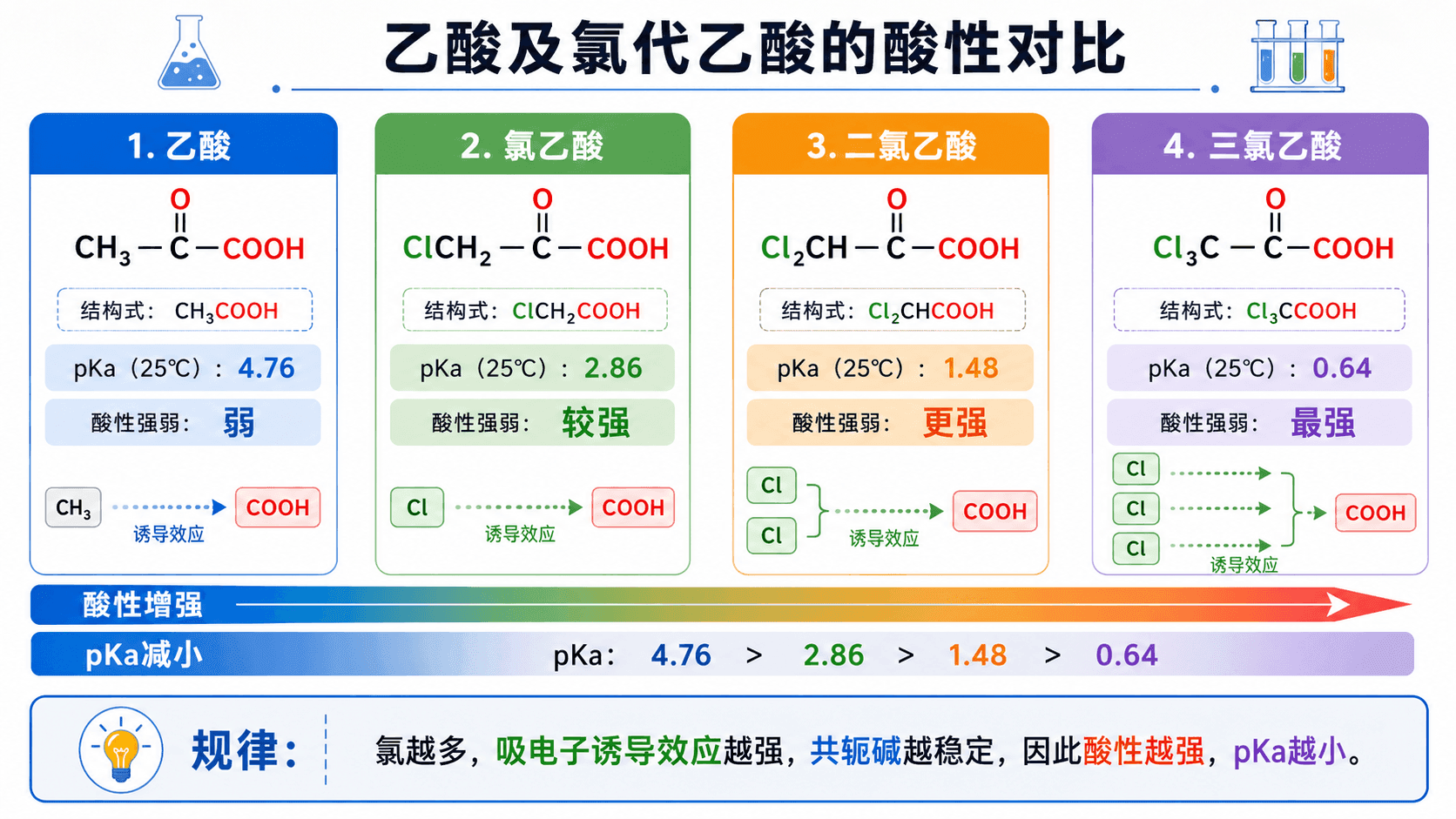
规律清晰可见:氯越多,吸电子诱导效应越强,酸性越强, 值越小。
例题:比较酸性强弱
例题: 将以下四种羧酸按照酸性从强到弱排列,并说明理由:
乙酸()、氟乙酸()、溴乙酸()、氯乙酸()
分析:
氟、氯、溴都是吸电子基团,连接在羧基 α 碳(紧邻羧基的碳)上,均增强酸性。吸电子能力强的使酸性越强,而卤素的吸电子能力(诱导效应)强弱顺序为:
因此酸性排序为:
结论: 氟乙酸酸性最强,乙酸酸性最弱,卤素的电负性越大,诱导效应越强,对酸性的增强作用越显著。
共轭效应:电子的“集体流动”
当电子不再“专属”于两个原子之间
诱导效应在单键体系中传递,且很快衰减。但有一类情况截然不同:当分子中存在交替出现的单键和多键(或含有孤对电子的原子),相邻的 p 轨道可以“肩并肩”地侧向重叠,电子云不再局限于两个特定原子之间,而是在更大的范围内流动、“共享”——这就是共轭效应(Conjugation Effect)。
π-π 共轭:双键与双键“手拉手”
1,3-丁二烯()是理解共轭效应的经典起点。
乍一看,这个分子中有两个孤立的 C=C 双键,应该和普通烯烃的行为类似。然而实验数据揭示了一个异常:1,3-丁二烯中间那根“单键”(C2—C3 键)的键长只有 147 pm,比普通 C—C 单键(154 pm)短了整整 7 pm;而两端的“双键”键长为 137 pm,比普通 C=C 双键(134 pm)略长。这说明两个双键之间发生了“相互渗透”——中间单键有了部分双键的性质,而两端双键也稍微“被分去”了一些电子。
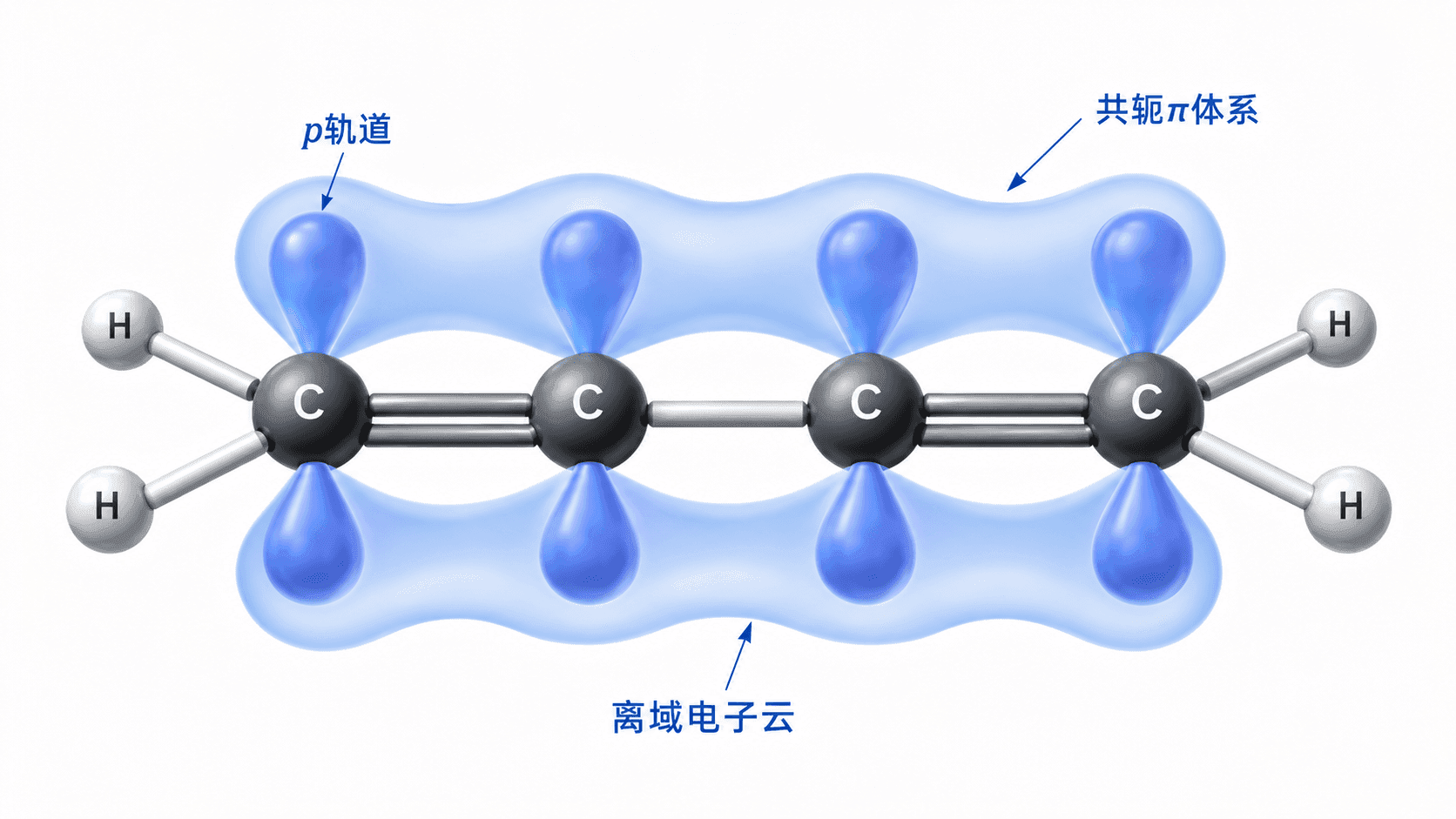
这正是 π-π 共轭的体现:两个 C=C 双键中各自含有的 π 键相互连通,形成了一个跨越 4 个碳的连续 π 体系,电子在其中“离域”分布。
离域后的体系能量比两个孤立双键更低——分子更稳定了。这额外的稳定化能量叫做共轭稳定化能(或离域能),1,3-丁二烯的共轭稳定化能约为 15 kJ/mol。
形成共轭的条件
并非任意两个双键都能形成共轭体系,需要满足以下三个条件:
-
第一, 相邻的 π 键或含有孤对电子的原子必须通过单键“间隔”相连,即双键-单键-双键交替出现(共轭链);
-
第二, 参与共轭的每个原子都需要有一个 p 轨道,且这些 p 轨道必须相互平行(共面);
-
第三, 各参与原子处于同一平面内(共轭体系一般为平面结构)。
判断是否形成共轭,关键在于检查双键之间的“间隔”。1,3-丁二烯中间只有一根单键,是共轭体系;而 1,4-戊二烯两个双键之间隔着两根单键,电子无法连续流通,属于孤立双键,不具有共轭效应。
p-π 共轭:孤对电子“加入”π 体系
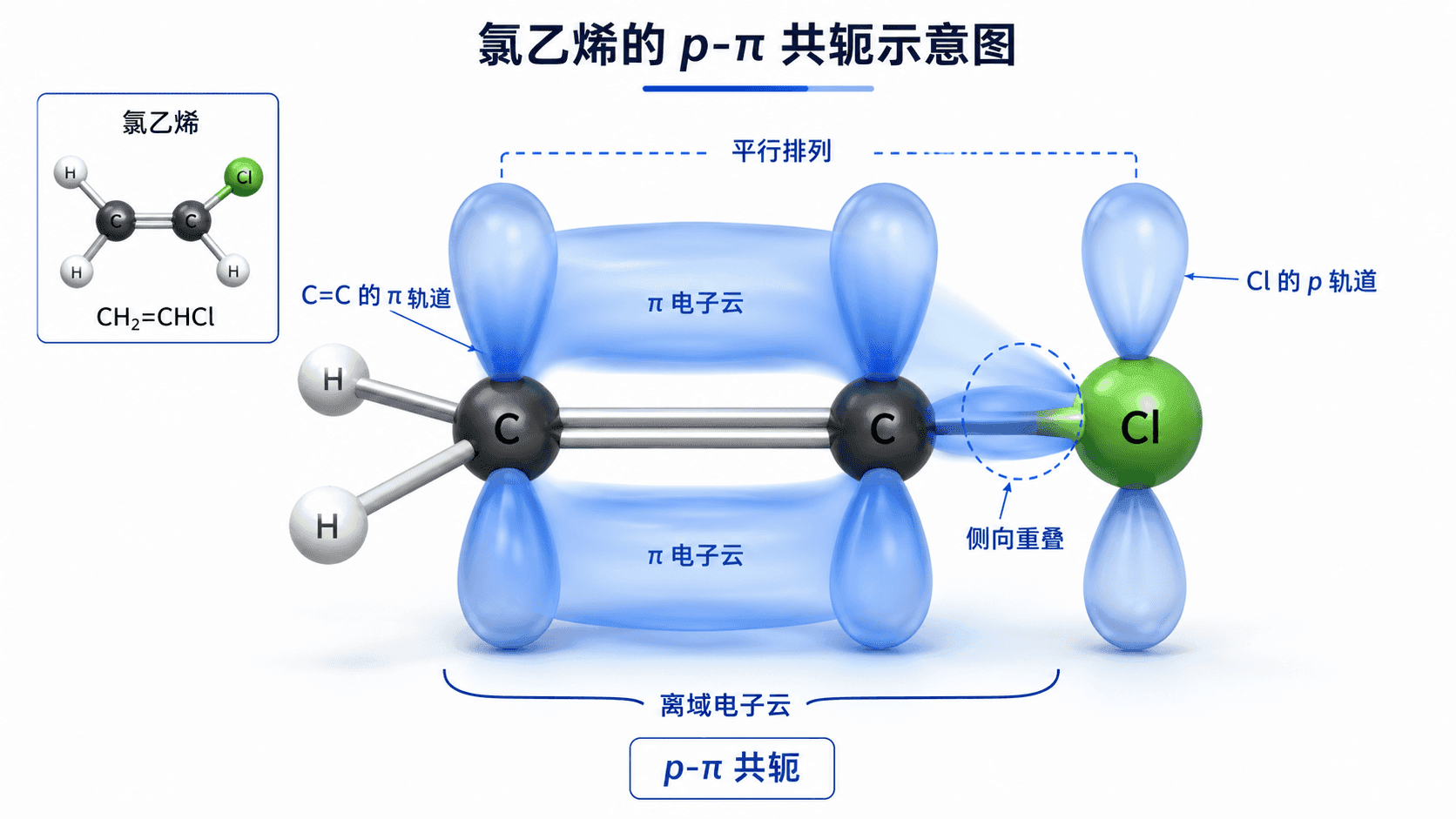
共轭效应不只发生在双键与双键之间,孤对电子也可以“掺和进来”,与相邻的 π 键形成 p-π 共轭。
氯乙烯()是最直观的例子。氯原子上有三对孤对电子,其中一对填充在与 C=C 的 π 轨道平行的 p 轨道中,从而与烯烃的 π 体系“连通”,形成 p-π 共轭。
实验结果印证了这一点:氯乙烯中 C—Cl 键的键长(172 pm)比氯乙烷()中的 C—Cl 键(179 pm)短,说明氯乙烯中 C—Cl 键有一定的双键性质——氯的孤对电子被部分“借”到了 π 体系中去。
同样的原理解释了苯胺()中,氨基(—)的碱性比普通胺弱的原因:氨基的氮原子孤对电子参与了苯环的 p-π 共轭,电子密度被“分散”到苯环上,氮原子上可以用来结合质子的电子密度降低,碱性因此减弱。
共轭效应的方向性
与诱导效应类似,共轭效应也有“推电子”和“拉电子”两个方向:
-
供电子共轭效应(+C 效应): 孤对电子向 π 体系推送,电子云密度增加。例如氯乙烯中 Cl 向双键推电子;苯胺中 — 向苯环推电子。
-
吸电子共轭效应(-C 效应): π 体系向另一个 π 键方向拉走电子,形成电子极性交替分布。例如 1,3-丁二烯中,双键之间的电子流动使得两端碳(C1、C4)带有相对较多的电子云,中间碳(C2、C3)稍少。
用箭头来表示共轭体系中的电子推移方向(弯箭头代表电子对的流动方向):
C1 和 C4 显示轻微负电性(),C2 和 C3 显示轻微正电性()——这正是 1,3-丁二烯与亲电试剂反应时,主要发生 1,4-加成(两端碳参与反应)的电子根源。
共轭效应的传递范围与诱导效应不同:诱导效应沿单键传递,3 根键后几乎消失;共轭效应则可以贯穿整个共轭链而基本不衰减,共轭链越长,电子离域范围越大,分子越稳定。
苯环——共轭体系的极致
苯()是共轭效应最典型的体现。苯环中 6 个碳原子各自提供一个 p 轨道,6 个 p 轨道彼此平行、形成闭合的环状离域π体系,6 个 π 电子“均匀分布”在整个苯环上,没有固定的单键与双键之分。
正因为如此:
苯的所有 C—C 键键长完全相等,均为 140 pm(介于单键 154 pm 和双键 134 pm 之间);
苯的氢化热(208 kJ/mol)远小于理论上三个孤立双键氢化热之和(3 × 119 = 357 kJ/mol),差值约 150 kJ/mol 正是苯环的共轭稳定化能。
这 150 kJ/mol 的“额外稳定性”使得苯不像普通烯烃那样容易发生加成反应,而是倾向于保留苯环结构发生取代反应,这也是后续讲解芳香取代反应的重要基础。
诱导效应与共轭效应并肩作战
两种效应的关键区别
在实际分子中,诱导效应和共轭效应往往同时存在,方向有时一致,有时相反。先清楚区分它们,是后续分析反应活性的基础。
卤苯中的“双重博弈”
氯苯()是同时存在两种效应的经典案例。
-
诱导效应层面: 氯的电负性(3.16)远大于碳(2.55),Cl 通过 C—Cl 的 σ 键“拉走”苯环上的电子,表现出 -I 效应,降低苯环的电子云密度。
-
共轭效应层面: 氯原子上的孤对电子,通过 p-π 共轭,向苯环 π 体系推送电子,表现出 +C 效应,增加苯环的电子云密度(尤其是邻、对位)。
两种效应方向相反:诱导效应吸电子,共轭效应供电子。在氯苯中,共轭效应(+C)比诱导效应(-I)略占上风,总体使苯环电子云密度略有增加,这也是为什么卤苯参与芳香取代反应时,取代基优先进入邻位和对位的根本原因。
电子效应与反应活性
两种效应最终归结到同一个问题上:“它使这个位置的电子多了还是少了?”
电子云密度增大的位置,更容易被缺电子的亲电试剂“攻击”;
电子云密度降低的位置,更容易被富电子的亲核试剂进攻。
用一个简单的框架整理:
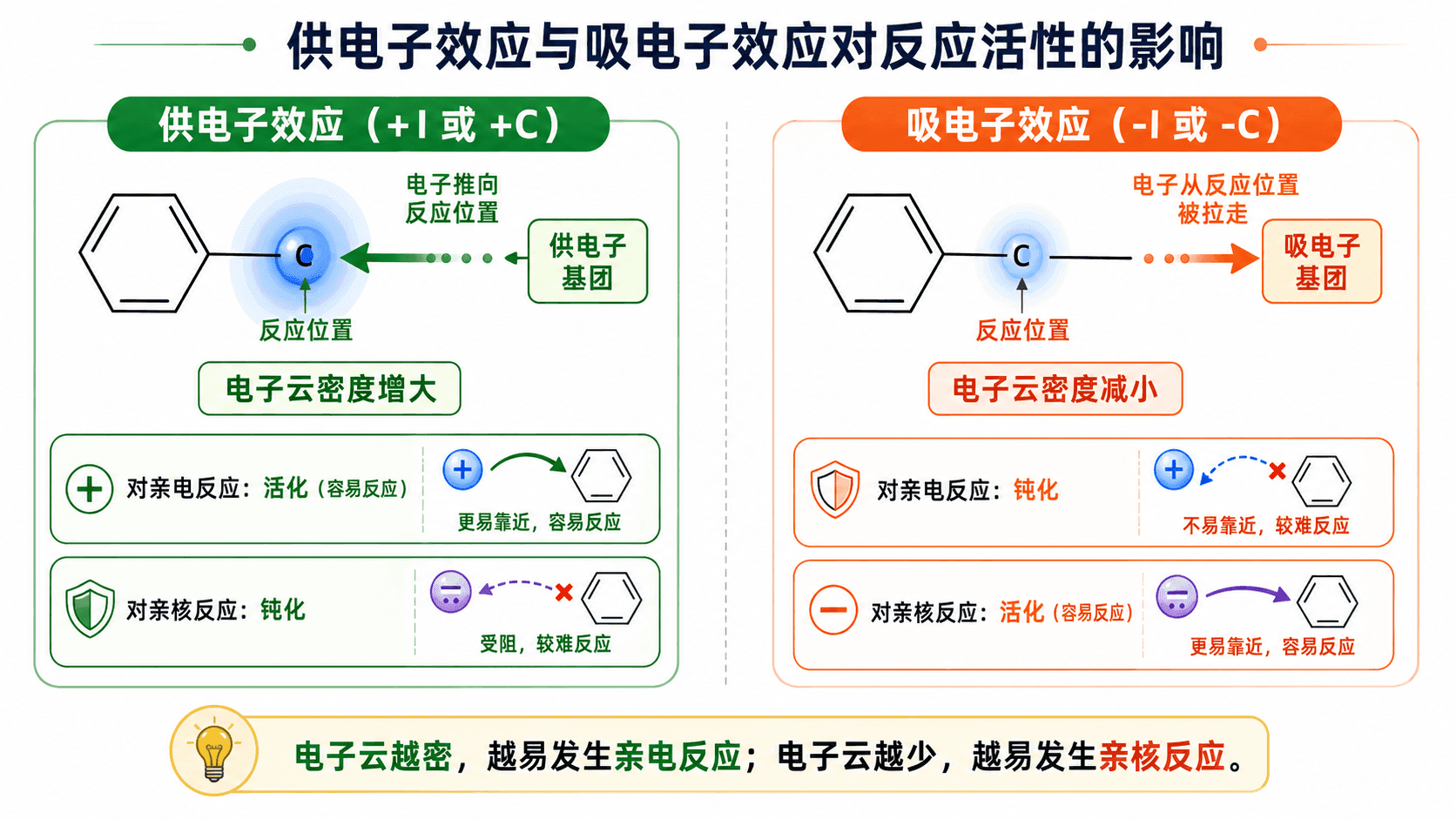
苯甲醛()中,醛基(—CHO)的 C=O 通过共轭效应和诱导效应双重吸电子,使苯环整体电子云密度降低,尤其是邻、对位降低更明显,因此苯甲醛在亲电取代中的活性低于苯,且取代主要发生在间位,这是后续讲取代规律时的重要例子。
例题:综合分析电子效应
例题: 对甲基苯甲酸(,又称对甲苯甲酸)与苯甲酸()相比,哪个酸性更强?请从电子效应的角度分析。
分析:
苯甲酸的酸性取决于羧酸根负离子()的稳定性以及苯环对羧基的电子影响。
对甲基苯甲酸在苯环对位引入了甲基()。甲基是供电子基团(+I 效应),通过诱导效应向苯环推送电子,使得苯环及羧基所在碳的电子云密度提高。羧基 O—H 键极性减小,氢更不容易以 离去;同时形成的羧酸根负离子因为甲基的推电子而更“不稳定”(负电荷更难分散),电离更困难。
实验数据证实:苯甲酸 ,对甲基苯甲酸 ,对甲基苯甲酸酸性确实更弱。
结论: 供电子基团(如烷基)降低羧酸酸性,吸电子基团(如卤素、硝基)增强羧酸酸性。
练习题
选择题
第 1 题【诱导效应方向判断】
下列基团中,属于吸电子诱导效应(-I 效应)基团的是:
A. (甲基)
B. (乙基)
C. (硝基)
D. (叔丁基)
答案:C
甲基、乙基、叔丁基均为烷基,电负性低于氢,属于供电子基团(+I 效应),A、B、D 错误。硝基(—)中氮连接两个电负性强的氧原子,整体表现出强吸电子诱导效应(-I 效应),选 C。
第 2 题【共轭体系判断】
下列分子中,存在 π-π 共轭体系的是:
A. 丙烷()
B. 1,4-戊二烯()
C. 1,3-丁二烯()
D. 乙烷()
答案:C
π-π 共轭需要两个双键之间仅隔一根单键(交替排列)。1,3-丁二烯的两个双键之间只有一根 C—C 单键,满足共轭条件,存在 π-π 共轭。1,4-戊二烯的两个双键之间隔了两根单键,属于孤立双键,不形成共轭。丙烷和乙烷均无双键,不存在共轭。
第 3 题【酸性强弱比较】
下列羧酸的酸性由强到弱排列正确的是:
A.
B.
C.
D.
答案:A
氯是强吸电子基团(-I 效应),氯原子数目越多,吸电子能力越强,羧酸酸性越强。因此酸性顺序为:三氯乙酸 > 二氯乙酸 > 氯乙酸 > 乙酸,选 A。
第 4 题【共轭效应的影响】
氯苯()中氯原子对苯环的综合电子效应是:
A. 仅有吸电子效应,使苯环电子云密度降低
B. 仅有供电子效应,使苯环电子云密度升高
C. 诱导效应吸电子(-I),共轭效应供电子(+C),总体使邻对位电子密度略升高
D. 诱导效应供电子(+I),共轭效应吸电子(-C),总体使苯环电子云密度降低
答案:C
氯的电负性大于碳,通过 σ 键表现出吸电子诱导效应(-I);同时氯原子的孤对电子通过 p-π 共轭向苯环推送电子,表现出供电子共轭效应(+C)。两者方向相反,在氯苯中 +C 效应略占优势,总体使苯环电子云密度略有增加,邻、对位尤为明显。
计算题
第 5 题【诱导效应与酸性——数据分析计算】
已知以下四种化合物的 数据(25°C,水溶液):
(1)计算氟乙酸相对乙酸的 差值 ,并换算成酸性常数 的比值(保留两位有效数字)。
(2)根据数据说明卤素诱导效应强弱顺序,并解释为什么卤乙酸均比乙酸酸性强。
解题过程:
(1)计算 和 比值:
降低 2.10,意味着:
即氟乙酸的酸性()约为乙酸的 130 倍。
(2)卤素诱导效应强弱分析:
从 数据可以看出:
越小,酸性越强,说明吸电子能力:,诱导效应由强到弱,与卤素电负性顺序一致(F > Cl > Br)。
卤乙酸均比乙酸酸性强,原因在于:卤素均为吸电子基团(-I 效应),通过 α 碳传递到羧基,使 O—H 键极性增大,氢更易以 形式离去;同时形成的羧酸根负离子因卤素继续抽取负电荷,负离子更稳定,促进电离平衡向右移动。
第 6 题【共轭稳定化能——氢化热数据应用】
氢化热是指每摩尔不饱和化合物完全氢化时放出的热量(取正值,单位 kJ/mol)。已知:
(1)若苯中三个双键是孤立的,预测其理论氢化热应为多少?
(2)将理论氢化热与实测值(208 kJ/mol)相比较,计算苯环的共轭稳定化能,并说明共轭效应如何影响苯的稳定性。
(3)1,3-环己二烯的氢化热(232 kJ/mol)小于两个孤立双键氢化热之和(2 × 120 = 240 kJ/mol),这说明了什么?
解题过程:
(1)苯的理论氢化热(假设三个孤立双键):
每个孤立 C=C 的氢化热以环己烯为参考:120 kJ/mol
(2)苯的共轭稳定化能:
苯的实测氢化热(208 kJ/mol)比理论值(360 kJ/mol)少了 152 kJ/mol,说明苯环因为完整的环状离域 π 体系,额外获得了约 152 kJ/mol 的共轭稳定化能。这使得苯环的能量远低于三个孤立双键的假想结构,化学上异常稳定,不容易发生破坏苯环的加成反应,而倾向于发生保留苯环结构的芳香亲电取代反应。
(3)1,3-环己二烯共轭稳定化的意义:
1,3-环己二烯的氢化热(232 kJ/mol)比两个孤立双键之和(240 kJ/mol)少 8 kJ/mol,说明 1,3-共轭二烯体系确实存在共轭稳定化效应,分子能量因电子离域而降低,但稳定化能(8 kJ/mol)远不如苯环(152 kJ/mol)——这正是开链共轭体系与闭合芳香 π 体系在稳定性上的本质差距。