热力学势与勒让德变换
热水壶烧开水、汽车发动机点火做功、化工厂里的合成反应——这些看似不同的过程,背后都有一个相同的问题:在给定条件下,系统最终会到达哪个状态,又能对外释放多少有用能量?内能 U U U
能量最小原理
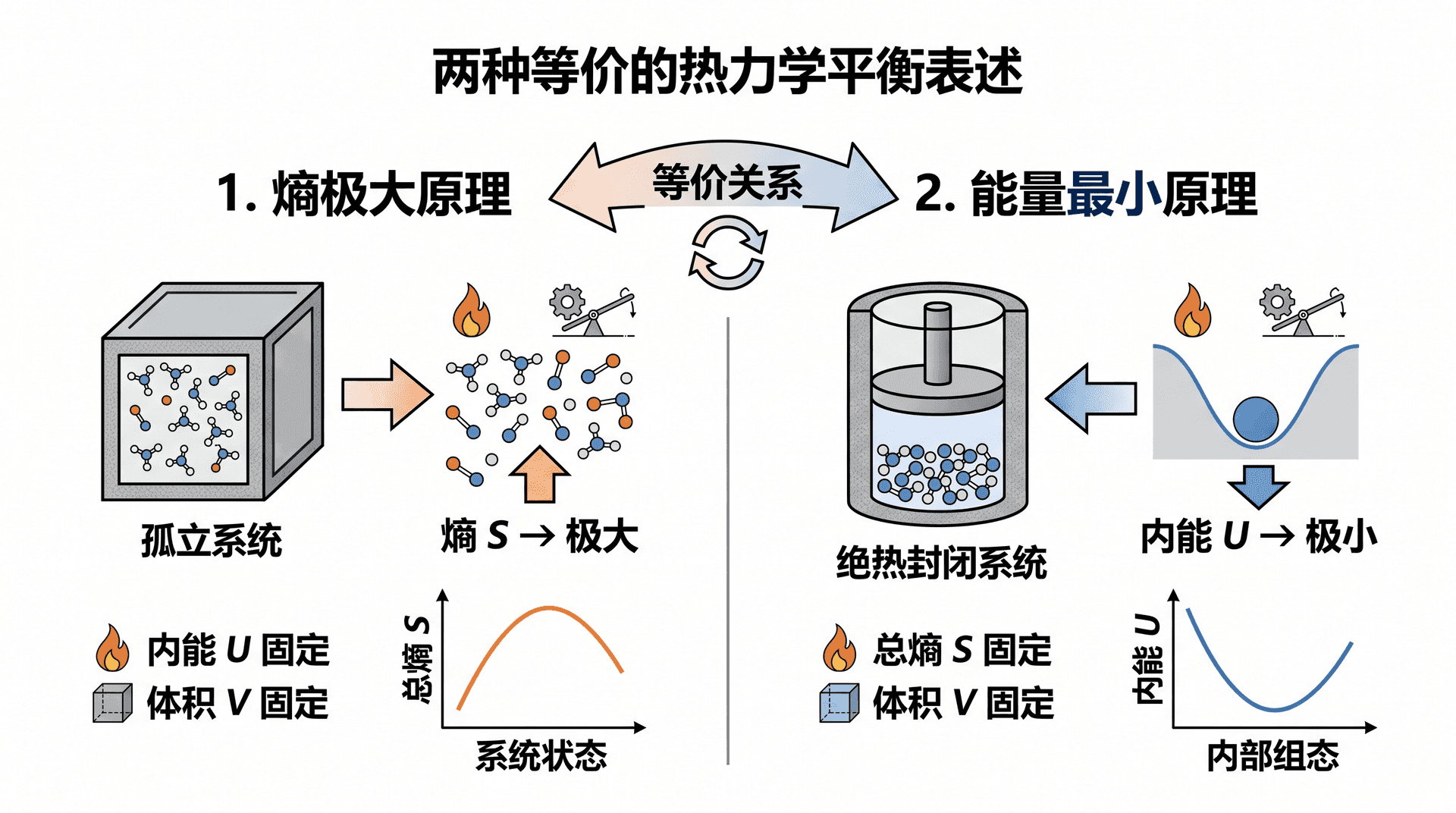
前面建立的熵极大假设指出:孤立系统达到平衡时,总熵取极大值。这一原理还有等价的能量表述,两者互为镜像,在不同场合各有用武之地。
将系统与外界的关系换一个视角来看:若固定系统的总熵 S S S U U U 能量最小原理 (Principle of Minimum Energy)。
用简单的逻辑可以理解这个原理:若在熵固定的情况下,系统还能通过某种内部重组使内能降低,那就意味着这份多余的能量可以释放出去做功,说明系统尚未达到平衡;当且仅当所有这样的可能都被穷尽,系统才真正处于平衡态。
例题 两个子系统 A A A B B B S = S A + S B S = S_A + S_B S = S A + S B U = U A + U B U = U_A + U_B U = U A + U B
内能对子系统分配方式求极小:令 d U = 0 dU = 0 d U = 0
d U A + d U B = 0 dU_A + dU_B = 0 d U A + d U B = 0 对每个子系统,利用热力学基本关系式 d U = T d S − P d V dU = T\,dS - P\,dV d U = T d S − P d V d V = 0 dV = 0 d V = 0 d S A + d S B = 0 dS_A + dS_B = 0 d S
T A d S A + T B d S B = 0 ⟹ ( T A − T B ) d S A = 0 T_A\,dS_A + T_B\,dS_B = 0 \implies (T_A - T_B)\,dS_A = 0 T A d S A + T B 因此 T A = T B T_A = T_B T A = T B
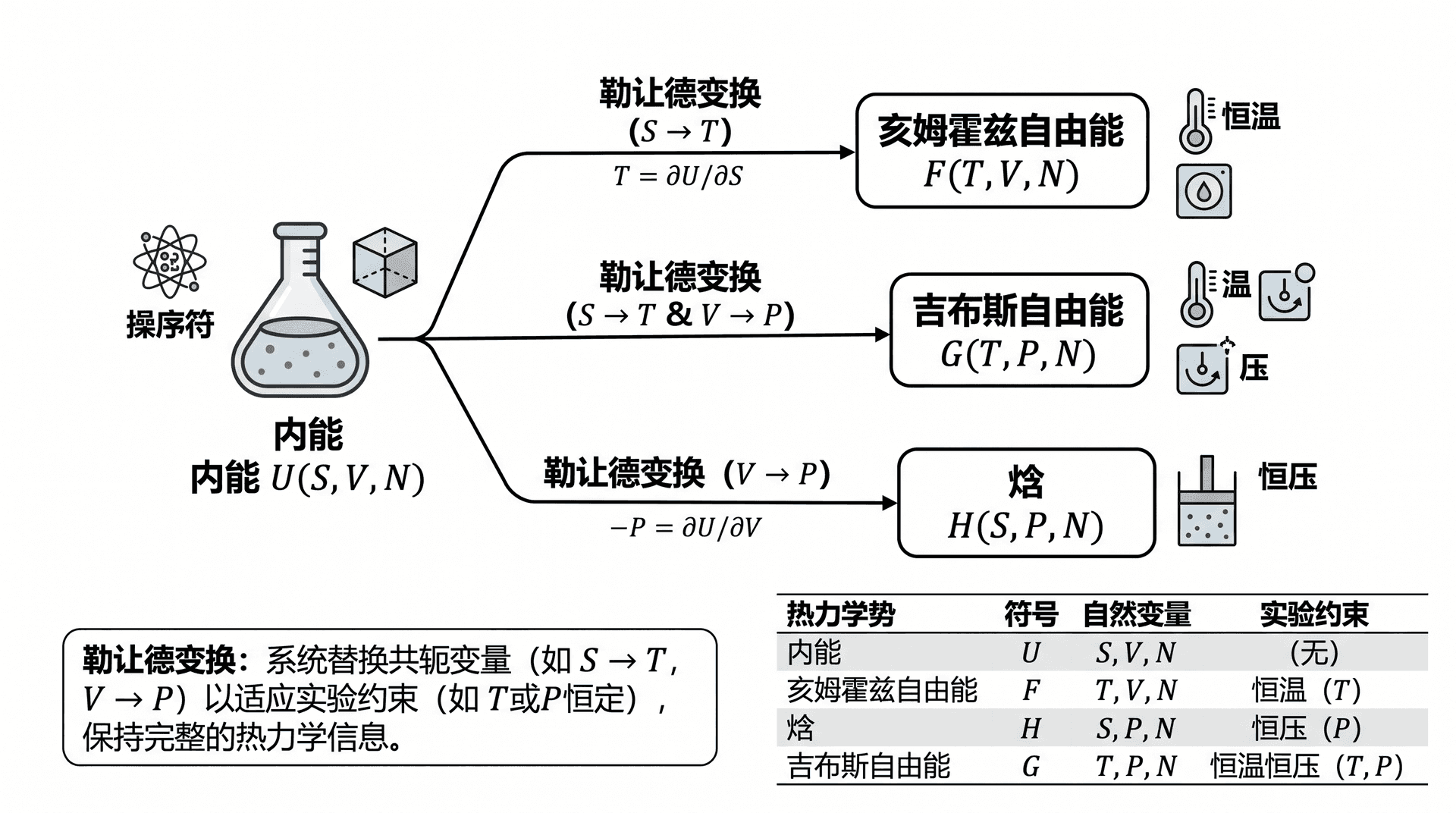
勒让德变换
内能 U = U ( S , V , N ) U = U(S, V, N) U = U ( S , V , N ) S S S V V V N N N T T T
数学上,若有函数 f ( x ) f(x) f ( x ) p = d f / d x p = df/dx p = df / d x
g ( p ) = f ( x ) − p x g(p) = f(x) - p\,x g ( p ) = f ( x ) − p x 在热力学中,将 U U U S S S T T T T = ∂ U / ∂ S T = \partial U/\partial S T = ∂ U / ∂ S V V V P P P
勒让德变换的精髓在于:变换前后的函数携带完全相同的热力学信息,只是「坐标系」不同。选择哪个热力学势,取决于实验的约束条件——让独立变量与约束条件一致,计算就会最为简洁。
四大热力学势
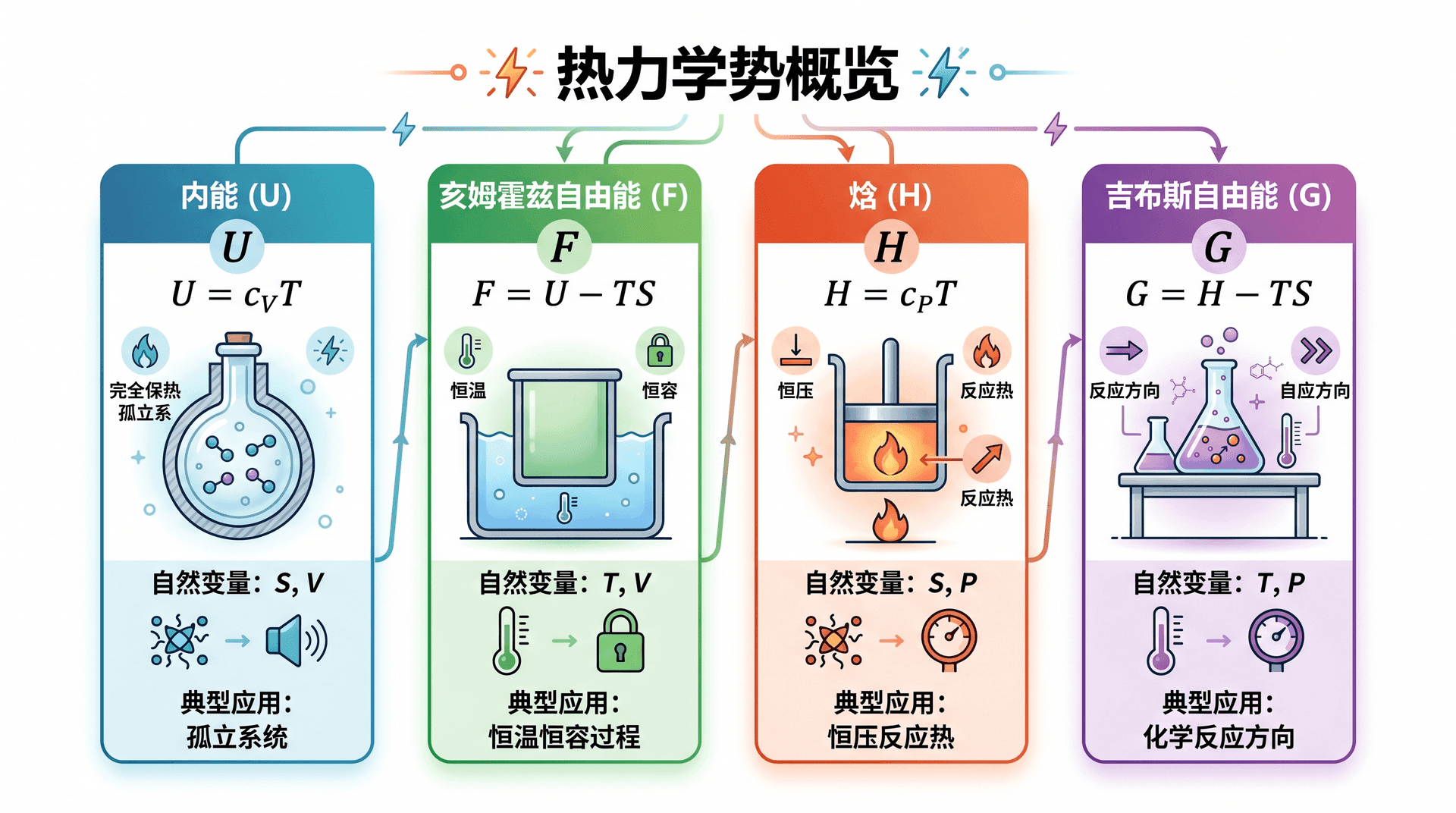
内能 U U U
内能是最基本的热力学势,自然变量为 S S S V V V N N N
d U = T d S − P d V + μ d N dU = T\,dS - P\,dV + \mu\,dN d U = T d S − P d V + μ d N 由此可以直接读出三个强度量:
T = ( ∂ U ∂ S ) V , N , P = − ( ∂ U ∂ V ) S , N , μ = ( ∂ U ∂ N ) S , V T = \left(\frac{\partial U}{\partial S}\right)_{V,N},\qquad P = -\left(\frac{\partial U}{\partial V}\right)_{S,N},\qquad \mu = \left(\frac{\partial U}{\partial N}\right)_{S,V} T = ( ∂ S ∂ U ) 亥姆霍兹自由能 F F F
对 S S S 亥姆霍兹自由能 (Helmholtz Free Energy):
F = U − T S F = U - TS F = U − T S 全微分为
d F = d U − T d S − S d T = − S d T − P d V + μ d N dF = dU - T\,dS - S\,dT = -S\,dT - P\,dV + \mu\,dN d F = d U − T d S − S d T = − S d T − P d V 自然变量变为 T T T V V V N N N
焓 H H H
对 V V V 焓 (Enthalpy):
H = U + P V H = U + PV H = U + P V 全微分为
d H = d U + P d V + V d P = T d S + V d P + μ d N dH = dU + P\,dV + V\,dP = T\,dS + V\,dP + \mu\,dN d H = d U + P d V + V d P = T d S + V d P + 自然变量为 S S S P P P N N N
吉布斯自由能 G G G
同时对 S S S V V V 吉布斯自由能 (Gibbs Free Energy):
G = U − T S + P V = F + P V = H − T S G = U - TS + PV = F + PV = H - TS G = U − T S + P V = F + P V = H − T S 全微分为
d G = − S d T + V d P + μ d N dG = -S\,dT + V\,dP + \mu\,dN d G = − S d T + V d P + μ d N 自然变量为 T T T P P P N N N
例题 写出 1 mol 1\ \text{mol} 1 mol U = c V T U = c_V T U = c V T P V = R T PV = RT P V = R T T T T
内能(以 T T T U 0 = 0 U_0 = 0 U 0 = 0
U = c V T U = c_V T U = c V T 亥姆霍兹自由能(利用理想气体熵 S = c V ln T − R ln P + 常数 S = c_V \ln T - R\ln P + \text{常数} S = c V ln T − R ln P + 常数
F = U − T S = c V T − T ( c V ln T − R ln P + s 0 ) F = U - TS = c_V T - T\bigl(c_V \ln T - R\ln P + s_0\bigr) F = U − T S = c V T − T ( c 焓(恒压过程,H = U + P V = c V T + R T = c P T H = U + PV = c_V T + RT = c_P T H = U + P V = c V T + R T = c P
H = c P T , c P = c V + R H = c_P T,\qquad c_P = c_V + R H = c P T , c P = c V + 吉布斯自由能:
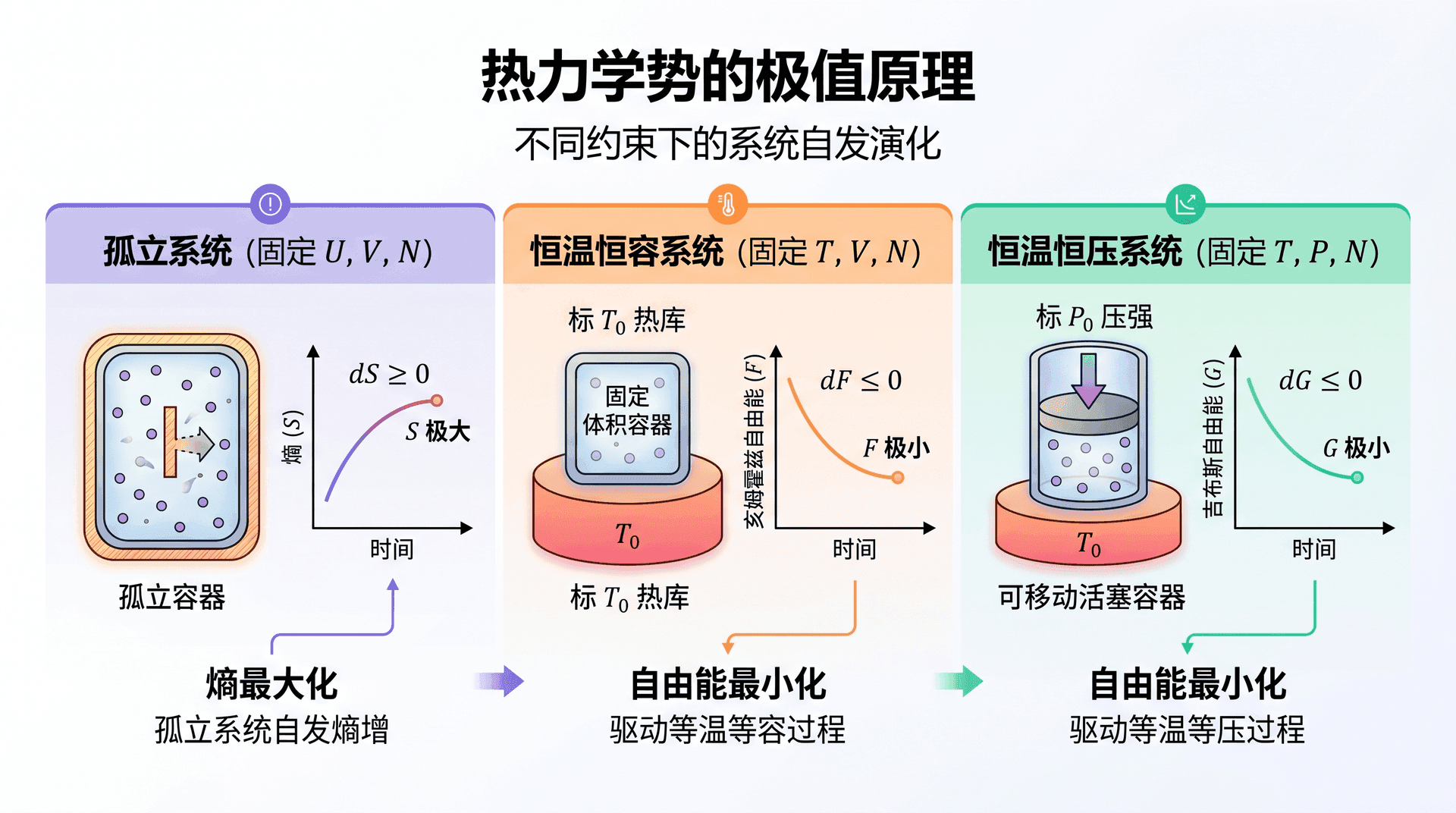
G = H − T S = c P T − T ( c P ln T − R ln P + s 0 ′ ) G = H - TS = c_P T - T\bigl(c_P \ln T - R\ln P + s_0'\bigr) G = H − T S = c P T − T ( c 热力学势的极值原理
不同热力学势各自在特定约束下取极小值,这是能量最小原理在不同「坐标系」下的体现。
恒温恒容系统(T T T V V V N N N
系统与温度为 T 0 T_0 T 0
d S 总 = d S 系 + d S 库 = d S − d U T 0 ≥ 0 dS_{\text{总}} = dS_{\text{系}} + dS_{\text{库}} = dS - \frac{dU}{T_0} \geq 0 d S 总 = d S 系 + d S 整理后得 d ( U − T 0 S ) ≤ 0 d(U - T_0 S) \leq 0 d ( U − T 0 S ) ≤ 0 d F ≤ 0 dF \leq 0 d F ≤ 0 F F F
恒温恒压系统(T T T P P P N N N
系统与温度为 T 0 T_0 T 0 P 0 P_0 P 0
d ( U − T 0 S + P 0 V ) ≤ 0 ⟹ d G ≤ 0 d(U - T_0 S + P_0 V) \leq 0 \implies dG \leq 0 d ( U − T 0 S + P 0 V ) ≤ 0 ⟹ 系统自发演化方向是吉布斯自由能减小,平衡态时 G G G
例题 1 mol 1\ \text{mol} 1 mol T = 300 K T = 300\ \text{K} T = 300 K A A A F A = − 1200 J F_A = -1200\ \text{J} F A = − 1200
Δ F = F B − F A = − 1350 − ( − 1200 ) = − 150 J < 0 \Delta F = F_B - F_A = -1350 - (-1200) = -150\ \text{J} < 0 Δ F = F B − F A = − 1350 − ( − 1200 Δ F < 0 \Delta F < 0 Δ F < 0 F F F
焓与节流过程
焓在恒压过程中有特别直观的含义。由 d H = T d S + V d P dH = T\,dS + V\,dP d H = T d S + V d P d P = 0 dP = 0 d P = 0 T d S T\,dS T d S δ Q rev \delta Q_{\text{rev}}
Δ H = Q P \Delta H = Q_P Δ H = Q P 恒压过程中的焓变等于系统吸收的热量。 这正是化学实验中用量热计测量反应热的理论基础。
节流过程(Joule-Thomson Process)是焓守恒的典型案例。气体在绝热管道中流过一个小孔(多孔塞或节流阀),两侧维持不同的压强 P 1 P_1 P 1 P 2 P_2 P 2
对穿过小孔的气体段做能量分析:上游对气体做功 P 1 V 1 P_1 V_1 P 1 V 1 P 2 V 2 P_2 V_2 P 2 V 2
U 2 − U 1 = P 1 V 1 − P 2 V 2 ⟹ U 2 + P 2 V 2 = U 1 + P 1 V 1 U_2 - U_1 = P_1 V_1 - P_2 V_2 \implies U_2 + P_2 V_2 = U_1 + P_1 V_1 U 2 − U 1 = P H 2 = H 1 \boxed{H_2 = H_1} H 2 = H 1 节流过程是等焓过程 (isenthalpic process)。气体经节流后压强降低,温度随之升高还是降低,取决于焦耳-汤姆孙系数 (Joule-Thomson Coefficient):
μ J T = ( ∂ T ∂ P ) H \mu_{JT} = \left(\frac{\partial T}{\partial P}\right)_H μ J T = ( ∂ P ∂ T ) 理想气体节流前后温度不变,因为其内能只与温度有关,等焓意味着等温。实际气体因为分子间存在相互作用,节流时分子间势能发生变化,温度才会改变。这一差异正是用节流效应来区分真实气体与理想气体行为的依据。
例题 氮气在室温(T 1 = 300 K T_1 = 300\ \text{K} T 1 = 300 K P 1 = 200 atm P_1 = 200\ \text{atm} P 1 = 200 atm P 2 = 1 atm P_2 = 1\ \text{atm}
节流过程是等焓过程,温度变化近似为
Δ T ≈ μ J T Δ P = 0.25 × ( 1 − 200 ) = 0.25 × ( − 199 ) ≈ − 49.8 K \Delta T \approx \mu_{JT}\,\Delta P = 0.25 \times (1 - 200) = 0.25 \times (-199) \approx -49.8\ \text{K} Δ T ≈ μ J T Δ P = 0.25 × ( 1 − 200 ) = T 2 ≈ 300 − 49.8 = 250.2 K T_2 \approx 300 - 49.8 = 250.2\ \text{K} T 2 ≈ 300 − 49.8 = 250.2 K 氮气经节流后温度下降约 50 K 50\ \text{K} 50 K 250 K 250\ \text{K} 250 K
吉布斯自由能与化学反应
吉布斯自由能是判断恒温恒压下化学反应能否自发进行的核心量。对于化学反应
反应物 ⟶ 产物 \text{反应物} \longrightarrow \text{产物} 反应物 ⟶ 产物 定义反应吉布斯自由能变 :
Δ G = G 产物 − G 反应物 \Delta G = G_{\text{产物}} - G_{\text{反应物}} Δ G = G 产物 − G 反应物 由 G = H − T S G = H - TS G = H − T S Δ G \Delta G Δ G
Δ G = Δ H − T Δ S \Delta G = \Delta H - T\Delta S Δ G = Δ H − T Δ S Δ G < 0 \Delta G < 0 Δ G < 0 Δ G = 0 \Delta G = 0 Δ G = 0 Δ G > 0 \Delta G > 0 Δ G > 0
例题 合成氨反应 N 2 + 3 H 2 → 2 NH 3 \text{N}_2 + 3\text{H}_2 \to 2\text{NH}_3 N 2 + 3 H 2 → 2 NH 3 T = 298 K T = 298\ \text{K}
在 T = 298 K T = 298\ \text{K} T = 298 K
Δ G = Δ H − T Δ S = − 92 000 − 298 × ( − 198 ) = − 92 000 + 59 004 = − 32 996 J/mol \Delta G = \Delta H - T\Delta S = -92\,000 - 298 \times (-198) = -92\,000 + 59\,004 = -32\,996\ \text{J/mol} Δ G = Δ H − T Δ S = − 92 000 − 298 × ( − 198 ) = − 92 Δ G ≈ − 33.0 kJ/mol < 0 \Delta G \approx -33.0\ \text{kJ/mol} < 0 Δ G ≈ − 33.0 kJ/mol < 0 反应在 298 K 298\ \text{K} 298 K Δ G = 0 \Delta G = 0 Δ G = 0
T ∗ = Δ H Δ S = − 92 000 − 198 ≈ 465 K T^* = \frac{\Delta H}{\Delta S} = \frac{-92\,000}{-198} \approx 465\ \text{K} T ∗ = Δ S Δ H = − 198 温度高于 465 K 465\ \text{K} 465 K Δ G > 0 \Delta G > 0 Δ G > 0
生成焓与焓的测量
焓的绝对值无法直接测量,实验中测量的总是焓变 Δ H \Delta H Δ H T = 298.15 K T = 298.15\ \text{K} T = 298.15 K P = 101.325 kPa P = 101.325\ \text{kPa} P = 101.325 kPa 1 mol 1\ \text{mol} 1 mol 标准摩尔生成焓 Δ f H ∘ \Delta_f H^\circ
利用赫斯定律 (Hess's Law),反应焓变等于产物生成焓之和减去反应物生成焓之和:
Δ r H ∘ = ∑ 产物 ν i Δ f H i ∘ − ∑ 反应物 ν j Δ f H j ∘ \Delta_r H^\circ = \sum_{\text{产物}} \nu_i\,\Delta_f H^\circ_i - \sum_{\text{反应物}} \nu_j\,\Delta_f H^\circ_j Δ r H ∘ = 产物 ∑ ν 其中 ν \nu ν
例题 计算乙醇完全燃烧的标准摩尔反应焓。
反应方程式:C 2 H 5 OH ( l ) + 3 O 2 ( g ) → 2 CO 2 ( g ) + 3 H 2 O ( l ) \text{C}_2\text{H}_5\text{OH}(l) + 3\text{O}_2(g) \to 2\text{CO}_2(g) + 3\text{H}_2\text{O}(l) C 2 H 5 OH ( l ) + 3 O 2
Δ r H ∘ = [ 2 × ( − 393.5 ) + 3 × ( − 285.8 ) ] − [ ( − 277.7 ) + 3 × 0 ] \Delta_r H^\circ = \bigl[2\times(-393.5) + 3\times(-285.8)\bigr] - \bigl[(-277.7) + 3\times 0\bigr] Δ r H ∘ = [ 2 × ( − 393.5 ) + = [ − 787.0 + ( − 857.4 ) ] − ( − 277.7 ) = − 1644.4 + 277.7 = − 1366.7 kJ/mol = \bigl[-787.0 + (-857.4)\bigr] - (-277.7) = -1644.4 + 277.7 = -1366.7\ \text{kJ/mol} = [ − 787.0 + ( − 857.4 ) ] − ( − 277.7 ) = − 1644.4 + 277.7 = − 1366.7 kJ/mol 每摩尔乙醇完全燃烧放热约 1367 kJ 1367\ \text{kJ} 1367 kJ Δ U \Delta U Δ U Δ H = Δ U + Δ ( P V ) ≈ Δ U + Δ n g R T \Delta H = \Delta U + \Delta(PV) \approx \Delta U + \Delta n_g RT Δ H = Δ U + Δ ( P V ) ≈ Δ U +
练习题
选择题
1. 在恒温恒压条件下,某化学反应的自发性由哪个热力学量的变化来判断?
A. 内能 U U U
B. 亥姆霍兹自由能 F F F
C. 吉布斯自由能 G G G
D. 熵 S S S
显示答案 答案:C
恒温恒压是实验室中最常见的化学反应条件,对应的热力学势是吉布斯自由能 G G G Δ G < 0 \Delta G < 0 Δ G < 0 S S S V V V
2. 下列关于焓的说法,正确的是
A. 焓是系统储存热量的多少,绝对值可以直接测量
B. 恒压可逆过程中,系统吸收的热量等于焓变 Δ H \Delta H Δ H
C. 节流过程中焓不守恒,因为存在压强变化
D. 理想气体节流后温度升高
显示答案 答案:B
由 d H = T d S + V d P dH = T\,dS + V\,dP d H = T d S + V d P d P = 0 dP = 0 d P = 0 d H = T d S = δ Q dH = T\,dS = \delta Q d H = T
3. 将内能 U ( S , V , N ) U(S, V, N) U ( S , V , N ) S S S
A. 焓 H = U + P V H = U + PV H = U + P V
B. 吉布斯自由能 G = U − T S + P V G = U - TS + PV G = U − T S + P V
C. 亥姆霍兹自由能 F = U − T S F = U - TS F = U − T S
D. 化学势 μ = ∂ U / ∂ N \mu = \partial U / \partial N μ = ∂ U / ∂ N
显示答案 答案:C
勒让德变换将独立变量 S S S T = ∂ U / ∂ S T = \partial U/\partial S T = ∂ U / ∂ S F = U − T S F = U - TS F = U − T S T , V , N T,\ V,\ N
4. 某反应在 T = 400 K T = 400\ \text{K} T = 400 K Δ H = + 60 kJ/mol \Delta H = +60\ \text{kJ/mol} Δ H = + 60 kJ/mol Δ S = + 200 J/(molK) \Delta S = +200\ \text{J/(molK)} Δ S = + 200 J/(molK)
A. 不能自发进行,因为 Δ H > 0 \Delta H > 0 Δ H > 0
B. 能自发进行,且 Δ G < 0 \Delta G < 0 Δ G < 0
C. 处于平衡状态,Δ G = 0 \Delta G = 0 Δ G = 0
D. 能否自发需要知道压强才能判断
显示答案 答案:B
Δ G = Δ H − T Δ S = 60 000 − 400 × 200 = 60 000 − 80 000 = − 20 000 J/mol < 0 \Delta G = \Delta H - T\Delta S = 60\,000 - 400 \times 200 = 60\,000 - 80\,000 = -20\,000\ \text{J/mol} < 0 Δ G = Δ H − T Δ S = 60 000 − 400 × 200 = 60 000 − 计算题
5. 某气体从高压端(P 1 = 100 atm P_1 = 100\ \text{atm} P 1 = 100 atm T 1 = 280 K T_1 = 280\ \text{K} T 1 = 280 K P 2 = 1 atm P_2 = 1\ \text{atm}
(1)求节流后的温度 T 2 T_2 T 2
(2)若改为可逆绝热膨胀(P 1 = 100 atm → P 2 = 1 atm P_1 = 100\ \text{atm} \to P_2 = 1\ \text{atm} P 1 = 100 atm → P 2 = 1 atm γ = C P / C V = 1.4 \gamma = C_P/C_V = 1.4
(3)比较两种膨胀方式的降温效果,说明哪种方式降温更显著。
显示答案 (1)节流后温度
节流过程等焓,温度变化为
Δ T ≈ μ J T ⋅ Δ P = 0.30 × ( 1 − 100 ) = 0.30 × ( − 99 ) = − 29.7 K \Delta T \approx \mu_{JT} \cdot \Delta P = 0.30 \times (1 - 100) = 0.30 \times (-99) = -29.7\ \text{K} Δ T ≈ μ J T ⋅ Δ P = 0.30 × ( 1 − 6. 工业合成甲醇的反应为 CO ( g ) + 2 H 2 ( g ) → CH 3 OH ( g ) \text{CO}(g) + 2\text{H}_2(g) \to \text{CH}_3\text{OH}(g) CO ( g ) + 2 H 2 ( g ) → CH 3 OH ( g ) 298.15 K 298.15\ \text{K}
(1)计算该反应在 298.15 K 298.15\ \text{K} 298.15 K Δ r H ∘ \Delta_r H^\circ Δ r H ∘
(2)计算标准摩尔反应熵 Δ r S ∘ \Delta_r S^\circ Δ r S ∘
(3)计算 298.15 K 298.15\ \text{K} 298.15 K Δ r G ∘ \Delta_r G^\circ Δ r G ∘
(4)估算使反应从自发变为不自发的临界温度 T ∗ T^* T ∗
显示答案 (1)标准摩尔反应焓
Δ r H ∘ = Δ f H ∘ ( CH 3 OH ) − [ Δ f H ∘ ( CO ) + 2 Δ f H ∘ ( H 2 ) ] \Delta_r H^\circ = \Delta_f H^\circ(\text{CH}_3\text{OH}) - \bigl[\Delta_f H^\circ(\text{CO}) + 2\Delta_f H^\circ(\text{H}_2)\bigr] Δ r H ∘ = Δ f