全基因组重测序
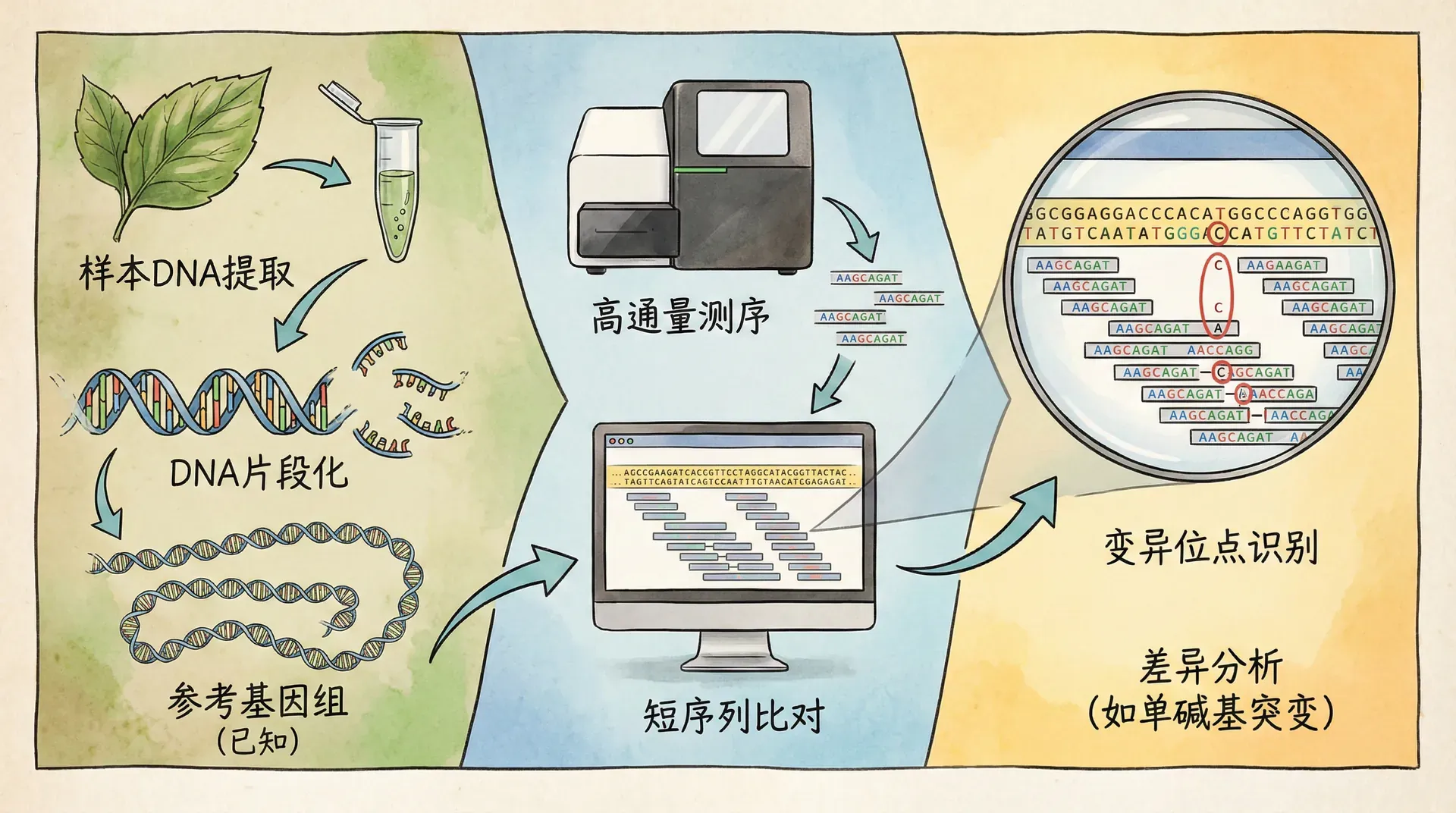
基因组重测序(Whole-genome resequencing, WGRS)是现代作物改良和进化研究的关键工具。它是在已有高质量参考基因组的基础上,对不同个体或品种进行全基因组测序,更高效地捕捉到SNP、InDel等遗传变异,也能检测到结构变异、拷贝数变化等复杂基因组重排,进而全面揭示物种内部的遗传多样性。
与传统测序相比,重测序不仅检测变异更准确高效,且适合大规模、多品种的系统分析,助力解析进化关系和筛选重要功能基因,为作物分子育种提供坚实基础。
全基因组重测序是在参考基因组基础上,对同一物种不同个体或群体进行高通量测序与比对,全面鉴定SNP、InDel、SV等遗传变异。该技术广泛应用于遗传多样性、驯化历史、性状基因挖掘和分子育种等研究。
技术发展历程
基因组测序技术的发展极大推动了生命科学,从最早的第一代Sanger测序,到革命性的第二代高通量测序(NGS),再到目前第三代单分子实时测序(如PacBio,Nanopore),测序通量和准确度经历飞跃式提升。中国在这一发展过程中不仅跟跑,更在很多领域开始领跑世界。例如,1999年,中国科学家参与了人类基因组计划,2003年华大基因主导完成了水稻基因组的测序。之后,二代测序技术的引入让测序成本从数百万美元降到千元人民币量级,催生了大规模重测序项目,包括对水稻、小麦、玉米、大豆等主要作物和许多野生植物及地方品种的群体测序。
下图展示了中国测序技术的发展历程、测序通量的提升以及行业重要节点:
中国科学家在作物基因组学领域取得了众多国际领先成果。不仅主导或参与水稻、小麦、玉米等作物基因组的测序与重测序项目,还推动测序技术自主创新。近年来,中国先后完成了水稻、小麦等高质量基因组,助力重要基因资源的发现与分子育种。相关成果不仅推动本土作物改良,也为全球农业科技进步和粮食安全贡献了“中国智慧”。
中国作物的进化历程探索
作物的驯化过程是一部贯穿数千年的宏大神话,凝结了人类智慧和自然选择的共同作用。从采集野生植物到有意识地选择和培育高产、抗逆、适口的作物,人类不断塑造着作物的进化轨迹。近十年来,随着全基因组重测序(Resequencing)等前沿技术的广泛应用,我们得以重建作物“家化”的完整分子历史,甚至动态捕捉到不同时空下基因与性状的共进化过程。这不仅丰富了我们对农作物起源、演化和地理扩散的认识,也为高效选育新品种提供了坚实理论基础。
水稻驯化

水稻是中国乃至全球最重要的粮食作物之一,其驯化历程可追溯至约9000年前的长江流域,成为中华农业文明的基石。早期的基因组学研究仅能揭示水稻驯化的大致过程,而随着高通量、高覆盖度全基因组测序数据的积累,驯化图景变得更加精细和复杂。
中国科学家依托对1,083个栽培稻品种和446个野生稻品种的全基因组重测序,发现粳稻和籼稻两大亚种呈现出彼此独立的驯化路径。现有研究表明,粳稻首先在中国南方的野生稻中被驯化为栽培稻,而籼稻的驯化可能与粳稻和野生稻的有性杂交密切相关。近年来,借助古DNA分析、地理起源追溯与功能验证等多手段,科学家更深入地解析了水稻驯化过程中的时空线路及关键遗传突变事件。除了粳稻和籼稻,杂交稻与具有特殊抗逆性的地方类型,也在驯化过程中展现出特定基因片段的选择与保留机制。
水稻主要亚种的驯化路径及特点:
水稻驯化的分子遗迹反映在产量、抗逆、品质等性状的持续提升上,同时也深刻影响了中国社会结构、生态环境与饮食文化。中国在全球水稻科学尤其是驯化机制研究中的贡献,不仅体现为基因测序数量与精度的世界领先,还包括数据库建设、功能注释、分子机制解析及研究标准输出,是作物驯化领域的国际领跑者。
主要作物的驯化和引种传播时间线
农作物的起源与扩散是地理环境、气候变化和人类选择共同作用的结果,中国是世界最重要的农作物驯化中心之一。不仅诞生了稻、粟、黍等世界性作物,也见证了小麦、玉米等外来作物的引入和本土改良。
让我们通过下表梳理中国主要作物的驯化和传播进程:
随着科学考古与分子生物学的结合,每一项农作物考古证据、每一组基因数据,正逐步拼出中国农业“进化树”的宏伟画卷。
通过基因组重测序技术,科学家们已发现58个与水稻驯化紧密相关的重要基因或功能位点。这些基因的变异和选择不仅帮助我们还原了作物家化的历史,更为分子育种、精准农业以及应对未来气候变化提供了丰富的靶点资源。
驯化过程中的基因组变革
农作物的驯化不仅仅是人工选择性状的积累过程,更伴随着大规模的基因组重组、结构变异及表观遗传调控。以水稻为例,科学家发现了一系列决定“野生”到“栽培”转变的关键性状相关基因。这些改变,从亚基因组的大片段丢失、基因拷贝数变化,到小尺度的SNP和Indel,无不深刻塑造了现代作物的性状谱系。
下方列举了水稻驯化过程中重要性状的变化及其功能基因:
此外,诸如开花期调控、株高、抗旱等性状的改良,也都溯源到全基因组重测序发现的关键基因。这些基因有的源于驯化前的野生种,有的源于驯化后選育的新品种。科学研究还显示,部分家化性状存在“选择瓶颈”,即在驯化和扩散过程中丢失了部分有益变异,因此现今的功能基因挖掘和农艺性状改良越来越重视野生种和地方品种的遗传资源。
发现作物的遗传宝库
中国以其广袤的国土和复杂的生态环境,孕育了丰富多样的农作物遗传资源。这些地方品种、野生近缘种和农家品种共同构成了中国作物的“基因库”,为未来作物改良、病虫害防控、产量与品质提升提供了取之不尽的分子基础。
中国作物品种的遗传多样性
以水稻为例,通过大规模全基因组重测序研究发现,中国地方水稻品种涵盖了全球水稻遗传多样性的70%以上。这一巨大遗传多样性,不仅体现在基因型、表型的丰富变异,也为应对气候变化、提升农产品多样性和稳定性提供了丰富材料。
事实上,中国不仅水稻,小麦、玉米、大豆、棉花等主要作物也都有庞大的地方品种库。科学家们常年对这些材料进行群体基因组分析,发现诸多具有抗逆、高产、优质等优势性状的新基因和功能位点。这些发现推动了中国特色的分子设计育种体系建设,有效支撑了国家粮食安全及农作物高质量发展。
从上图可以看到,在所有主要作物中,地方品种的遗传多样性显著高于现代商业品种。这种资源的积累和保存提升了中国应对农作物病虫害、极端气候及粮食安全挑战的能力。
分子标记技术的应用
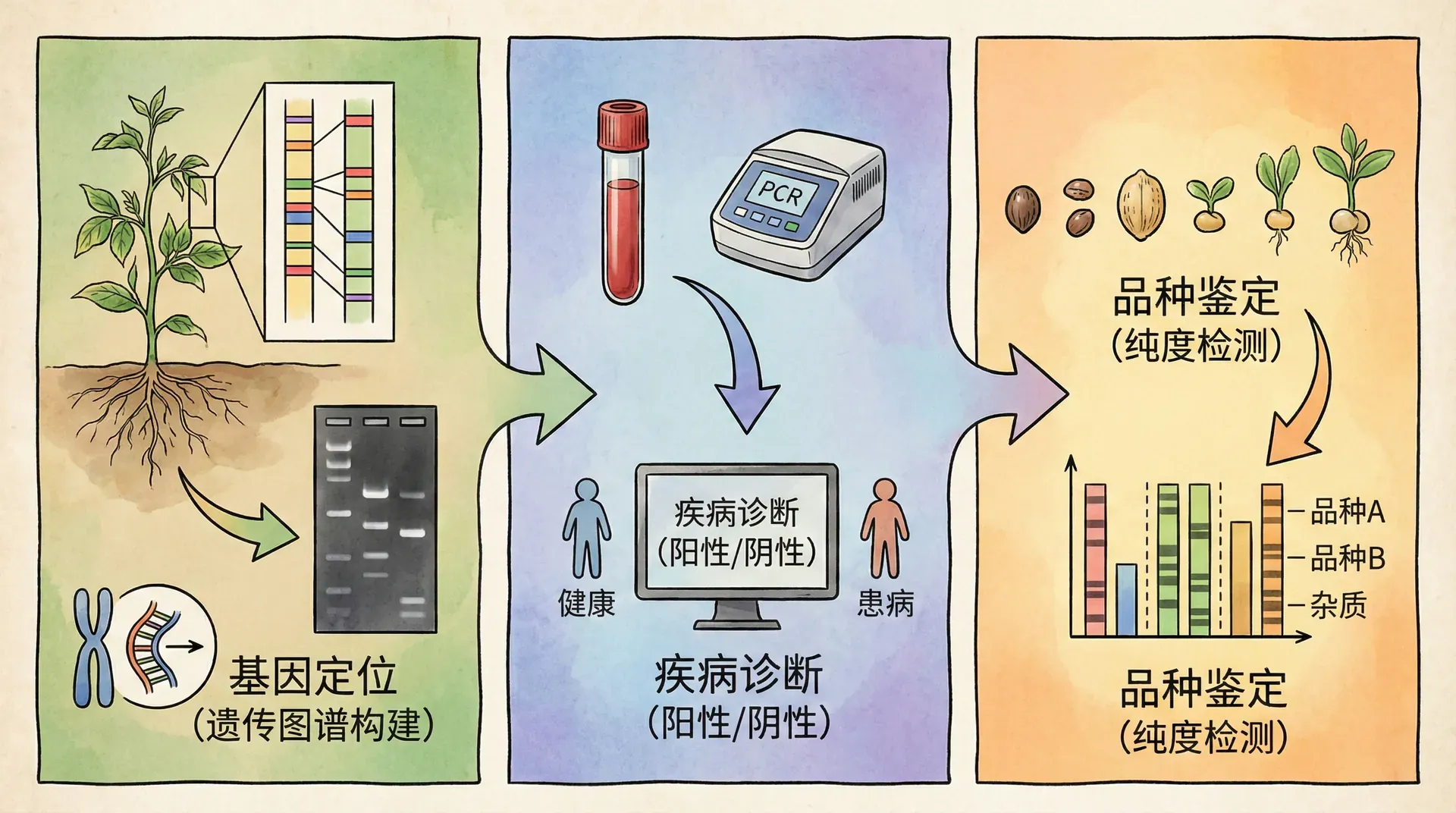
全基因组重测序不仅揭示了作物进化与多样性的本质,还极大拓展了分子标记的发现与开发能力。最具代表性的分子标记是单核苷酸多态性(SNP),近年来还包括结构变异(SV)、InDel等多种类型。这些标记如同基因组上的“路标”或“导航仪”,能精准帮助科学家定位调控农艺性状的功能区域。
现代全基因组重测序实验一次即可发现数百万个SNP标记,极大提升了群体遗传学、遗传连锁分析、QTL定位和分子育种的效率,为传统方法带来革命性升级。
中国科学家广泛应用这些标记技术,推动了作物分子辅助育种、优良基因导入及全基因选择育种等一系列创新工程,取得了诸多突破性成果。例如:
随着第三代测序和人工智能分析等新技术加持,分子标记开发和应用的数量与精度还将持续提升,为中国乃至世界农业科技进步不断注入新动能。
寻找重要农艺性状基因
基因的发现与功能解析一直是作物遗传改良研究的核心内容。可以将基因发现过程形象地比喻为在浩瀚无垠的基因组“海洋”中撒网捞珍珠——每一个重要性状基因的发现,都能极大推动农业生产力的提升和作物品种的改良。
近年来,随着全基因组重测序(WGS)等高通量组学技术的发展,科学家们可以以前所未有的分辨率和速度从庞大的自然变异资源库中寻找与农艺性状紧密相关的关键基因。无论是产量、品质,还是抗逆性、抗病性等复杂性状的遗传基础,正被逐步揭开面纱。全基因组关联分析(GWAS)正是当今国际主流的基因挖掘手段之一。
全基因组关联分析原理
GWAS(Genome-Wide Association Study)是一种基于自然群体遗传变异和表型变异联合分析的遗传解析方法。其基本原理是利用全基因组范围内的高密度分子标记(如SNP),在大规模的自然群体中,通过统计学方法寻找与目标性状显著关联的基因组区域(即QTL或候选基因)。
这类似于在浩繁的数据中运用“大数据挖掘+精细定位”技术,揭示遗传变异与表型变异之间的因果关系。GWAS可以定位到具体的染色体区段或候选基因,极大提升基因克隆和功能鉴定的效率。
成功的GWAS分析需要高质量的群体基因型数据和准确的表型测定结果,这往往依赖于全基因组重测序技术带来的高密度标记和更多的自然变异信息。
GWAS分析过程简述
- 群体构建:选择足够数量和代表性强的自然品种或驯化材料,保证表型多样性和遗传变异丰富。
- 基因型检测:采用全基因组重测序、芯片等方法获得高密度的SNP或InDel分子标记信息。
- 表型鉴定:对目标性状进行多环境、多指标的精准测定,积累高质量表型数据。
- 关联分析:利用统计和生信方法(如混合线性模型),关联基因型与表型,筛选出显著关联的基因组区域。
- 后续功能验证:对关联信号强的候选基因进行分子克隆、表达验证或基因编辑,明确功能机制。
GWAS分析结果可视化
GWAS的常见结果展示手段是“曼哈顿图”,以染色体物理位置为横轴、关联显著性(通常为-log10(P值))为纵轴,高峰代表显著关联区域。如下表以曼哈顿图数据格式列举部分显著关联点:
通过曼哈顿图,研究者能直观地发现与目标性状最显著相关的位点,优先开展后续的定位与功能分析。
中国科学家的基因发现成果
中国科学家在重要作物基因发现与功能解析方面已取得大批突破性进展,并产生了世界级影响。例如:
中国科学家还积极开展功能基因网络解析、重要基因库构建、大规模基因编辑与分子设计育种,为全球农作物基因资源的发掘和利用做出了重要贡献。
基因发现的实际应用价值
这些基因的发现不仅丰富了对作物性状遗传机理的科学认知,更直接服务于现代育种实践,成为品种选育与优化的重要利器。通过基于分子标记的辅助选择(MAS),特定性状基因可以被有效追踪,大大提高育种的效率,实现“精准选材”。
通过对比可以发现,应用分子标记辅助选择(MAS),目标性状的改良速度和幅度明显提升。
分子标记辅助选择能够将新品种的育种周期缩短2-3倍,大大加快了优质品种的选育和应用步伐,同时有效提高选育的准确性和可控性。
此外,现代全基因组重测序结合分子标记开发,还推动了基因组选择(GS)等新兴育种技术的发展,使得全基因组范围的遗传潜力能够被系统性利用。未来的大数据、人工智能与基因编辑的深度整合,将进一步加速基因发现、标记开发与高效育种的实践落地。
综上所述,全基因组重测序技术极大丰富了作物重要性状基因的发现渠道和分子资源库,实现了从基因解析到精准改良的全链条创新。它不仅为中国农业科技自立自强、粮食安全和可持续发展提供了坚实基础,也为世界作物基因组学与现代育种技术提供了“中国方案”。
将来随着测序技术更加普惠、人工智能分析能力提高与多组学信息的融合,全基因组重测序与基因发掘将在作物遗传改良、种质资源创新和应对全球粮食安全挑战中持续发挥更为核心的作用。